The Laboratory of Microglial Phagocytosis,
Neuroinflammation & Neurodegeneration
The Laboratory of Guy Brown at the University of Cambridge
Home - Publications - News - Biography of Guy Brown - Lab members - Contact
Latest lab publications
Microglial phagocytosis in Alzheimer disease
News summary: Microglial phagocytosis is the engulfment of extracellular things by microglia, which are brain macrophages. We review the evidence here that microglial phagocytosis is central to Alzheimer disease.

Microglial phagocytosis in Alzheimer Disease
Abstract:
Accumulating evidence indicates that Alzheimer disease (AD) is caused by dysregulated microglial phagocytosis. The main risk factor for
AD is age, and ageing reduces microglial phagocytosis of amyloid-β (Aβ) plaques, while increasing microglial phagocytosis of synapses and neurons. Most of the known genetic risk for AD can be linked to microglial phagocytosis, including ABCA1, ABI3, ACE, ADAM17, APOE, APP, BIN1, BLNK, CD2AP, CD33, CLU, CR1, CTSB, CTSH, EED, GRN, INPP5D, LILRB2, PICALM, PLCG2, PSEN1, PTK2B, SIGLEC11, SORL1, SPI1, TMEM106B and TREM2. Moreover, the only disease-modifying treatments for AD — anti-Aβ antibodies — work by increasing microglial phagocytosis of Aβ aggregates. Microglial phagocytosis of Aβ via TREM2, LRP1, CD33, TAM receptors and anti-Aβ antibodies appears to reduce AD pathology by pruning and compacting plaques, restricting subsequent tau pathology, whereas microglial phagocytosis of synapses and neurons seems detrimental in the later stages of AD, via complement, P2Y6 receptor and TREM2. However, the roles of microglial phagocytosis in AD are complex and multifaceted, and improved treatments are likely to require a deeper understanding of these roles.
Brown, G.S., St George-Hyslop, P., Paolicelli, R.C. & Lemke, G. (2025) Microglial Phagocytosis in Alzheimer Disease. Nature Reviews Neurology.
Why things look the way they do:
we live in a Cartesian grid of reified concepts
News summary: The human world around us is becoming increasingly conceptual, because we are externalising concepts into the environment as objects. And these objects are designed to evoke concepts in our minds. Non-conceptual material is actively removed from human environments as trash, giving the illusion that reality is conceptual.

The Ames illusion (above) illustrates how the Cartesian grid of a ‘normal’ room helps judge the depth, shape and position of objects.
Abstract:
The world we live in now is almost exclusively populated by things designed by minds to be understood by other minds. I argue here that: (1) Human environments now consist mainly of reified concepts, such as “chairs.” (2) These externalized concepts look like simple cartoons of the concepts they reify, with flat, homogenous surfaces in geometric shapes, with one or a small number of colors, textures and surfaces, so that they can be easily identified and distinguished. (3) Reified concepts are organized within a Cartesian grid, that enables their perception, location and memory. (4) Simple concepts are nested within complex concepts, such as rooms, houses, streets, hospitals or cities, designed to be read by our minds, and guide behavior, within the externalized mind of society. (5) Components of the human environment that are not conceptual are actively removed, resulting in a very low entropy of information, and giving the illusion that reality is entirely conceptual. (6) Reified concepts and their conceptual ordering help perception, comprehension and use of human environments. (7) These ideas have application to design, architecture, aesthetics, phenomenology, ontology and understanding why things look the way they do.
Brown GC (2025) Why things look the way they do: we live in a Cartesian grid of reified concepts. Front. Psychol. 16:1612191.
S100A9 protein activates microglia and stimulates phagocytosis, resulting in synaptic and neuronal loss
News Summary
S100A9 is a protein with a much increased concentration in the brains of Alzheimer’s disease patients. We find that S100A9 aggregates and induces loss of synapses and neurons in co-cultures of neurons and glial cells. S100A9 induce microglial proliferation, activation and pro-inflammatory cytokine production. And the.neuronal loss induced by S100A9 is prevented if microglia are removed. Thus, S100A9 appears to cause neurodegeneration by activating microglia to phagocytose synapses and neurons.
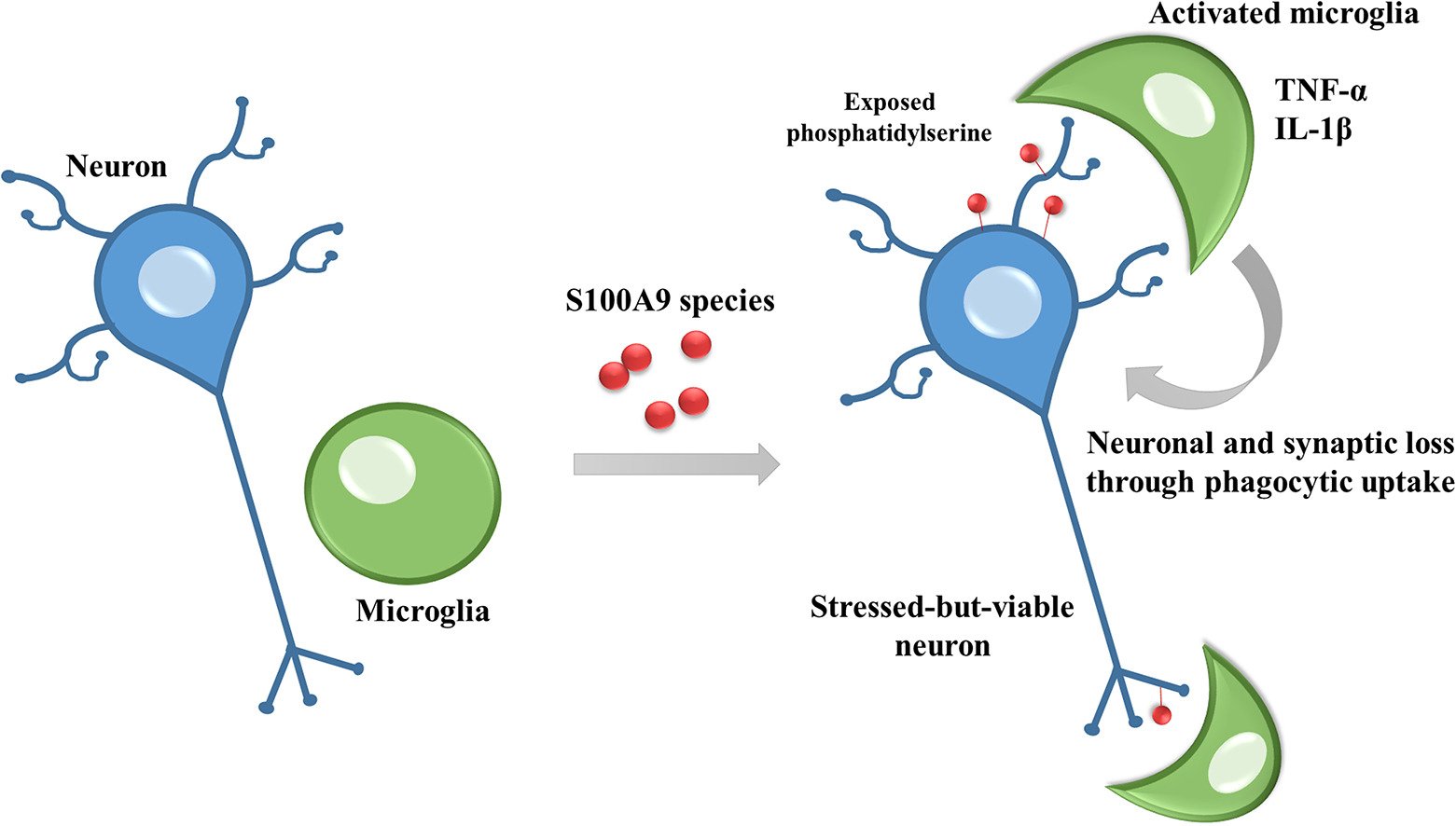
Abstract
Myelin is required for the function of neuronal axons in the central nervous system, but the mechanisms that support myelin health are unclear. Although macrophages in the central nervous system have been implicated in myelin health1, it is unknown which macrophage populations are involved and which aspects they influence. Here we show that resident microglia are crucial for the maintenance of myelin health in adulthood in both mice and humans. We demonstrate that microglia are dispensable for developmental myelin ensheathment. However, they are required for subsequent regulation of myelin growth and associated cognitive function, and for preservation of myelin integrity by preventing its degeneration. We show that loss of myelin health due to the absence of microglia is associated with the appearance of a myelinating oligodendrocyte state with altered lipid metabolism. Moreover, this mechanism is regulated through disruption of the TGFβ1-TGFβR1 axis. Our findings highlight microglia as promising therapeutic targets for conditions in which myelin growth and integrity are dysregulated, such as in ageing and neurodegenerative disease.
Pampuscenko K, Jankeviciute S, Morkuniene R, Sulskis D, Smirnovas V, Brown GC, Borutaite V. S100A9 protein activates microglia and stimulates phagocytosis, resulting in synaptic and neuronal loss. Neurobiol Dis. 2025 Jan 28:106817.
Neuroinflammation in Alzheimer disease
Heneka MT, van der Flier WM, Jessen F, Hoozemanns J, Thal DR, Boche D, Brosseron F, Teunissen C, Zetterberg H, Jacobs AH, Edison P, Ramirez A, Cruchaga C, Lambert JC, Laza AR, Sanchez-Mut JV, Fischer A, Castro-Gomez S, Stein TD, Kleineidam L, Wagner M, Neher JJ, Cunningham C, Singhrao SK, Prinz M, Glass CK, Schlachetzki JCM, Butovsky O, Kleemann K, De Jaeger PL, Scheiblich H, Brown GC, Landreth G, Moutinho M, Grutzendler J, Gomez-Nicola D, McManus RM, Andreasson K, Ising C, Karabag D, Baker DJ, Liddelow SA, Verkhratsky A, Tansey M, Monsonego A, Aigner L, Dorothée G, Nave KA, Simons M, Constantin G, Rosenzweig N, Pascual A, Petzold GC, Kipnis J, Venegas C, Colonna M, Walter J, Tenner AJ, O'Banion MK, Steinert JR, Feinstein DL, Sastre M, Bhaskar K, Hong S, Schafer DP, Golde T, Ransohoff RM, Morgan D, Breitner J, Mancuso R, Riechers SP. Neuroinflammation in Alzheimer disease. Nat Rev Immunol. 2024 Dec 9. doi: 10.1038/s41577-024-01104-7.
Neuroinflammation in Alzheimer disease. Nat Rev Immunol (2024).
Abstract
Increasing evidence points to a pivotal role of immune processes in the pathogenesis of Alzheimer disease, which is the most prevalent neurodegenerative and dementia-causing disease of our time. Multiple lines of information provided by experimental, epidemiological, neuropathological and genetic studies suggest a pathological role for innate and adaptive immune activation in this disease. Here, we review the cell types and pathological mechanisms involved in disease development as well as the influence of genetics and lifestyle factors. Given the decade-long preclinical stage of Alzheimer disease, these mechanisms and their interactions are driving forces behind the spread and progression of the disease. The identification of treatment opportunities will require a precise understanding of the cells and mechanisms involved as well as a clear definition of their temporal and topographical nature. We will also discuss new therapeutic strategies for targeting neuroinflammation, which are now entering the clinic and showing promise for patients.
Neuroinflammation in Alzheimer disease. Nat Rev Immunol (2024).
The links between neuroinflammation, brain structure and depressive disorder: A cross-sectional study protocol
News Summary
This paper describes the protocol of a study of the relationship between: i) inflammogens in blood (LPS and cytokines), ii) brain structure measured by MRI, and iii) clinical depression.
Milasauskiene E, Burkauskas J, Jesmanas S, Gleizniene R, Borutaite V, Skemiene K, Vaitkiene P, Adomaitiene V, Lukosevicius S, Gradauskiene B, Brown G, Steibliene V. The links between neuroinflammation, brain structure and depressive disorder: A cross-sectional study protocol. PLoS One. 2024 Nov 20;19(11):e0311218.
Abstract
Introduction: It is known that symptoms of major depressive disorder (MDD) are associated with neurodegeneration, that lipopolysaccharide (LPS) can induce symptoms of MDD, and that blood LPS levels are elevated in neurodegeneration. However, it is not known whether blood LPS and cytokine levels correlate with MDD, cognition and brain structure, and this is tested in this study.
Methods and analysis: This cross-sectional study includes individuals with MDD (n = 100) and a control group of individuals with no one-year history of a mental disorder (n = 50). A comprehensive evaluation is performed, including the collection of basic sociodemographic information, data on smoking status, body mass index, course of MDD, past treatment, comorbid diseases, and current use of medications. Diagnosis of MDD is performed according to the WHO's [2019] International Classification of Diseases and related health problems by psychiatrist and severity of MDD is evaluated using the Montgomery-Åsberg Depression Scale. The Cambridge Neuropsychological Test Automated Battery is used to evaluate cognitive functioning. Venous blood samples are taken to measure genetic and inflammatory markers, and multiparametric brain magnetic resonance imaging is performed to evaluate for blood-brain barrier permeability, structural and neurometabolic brain changes. Descriptive and inferential statistics, including linear and logistic regression, will be used to analyse relationships between blood plasma LPS and inflammatory cytokine concentrations in MDD patients and controls. The proposed sample sizes are suitable for identifying significant differences between the groups, according to a power analysis.
Administrative information: Trial registration: Clinicaltrials.gov NCT06203015.
The microglial P2Y6 receptor as a therapeutic target for neurodegenerative diseases
News Summary: The P2Y6 receptor is a receptor on the surface of microglia (brain macrophages) that stimulates microglial phagocytosis and inflammation when activated by extracellular uridine diphosphate. We review here the evidence that inhibition of this receptor can reduce neurodegeneration and other brain pathologies modelled in mice. Thus the P2Y6 receptor is a promising therapeutic target to treat Alzheimer’s disease and other brain pathologies that are mediated by microglial inflammation and phagocytosis of synapses and neurons.
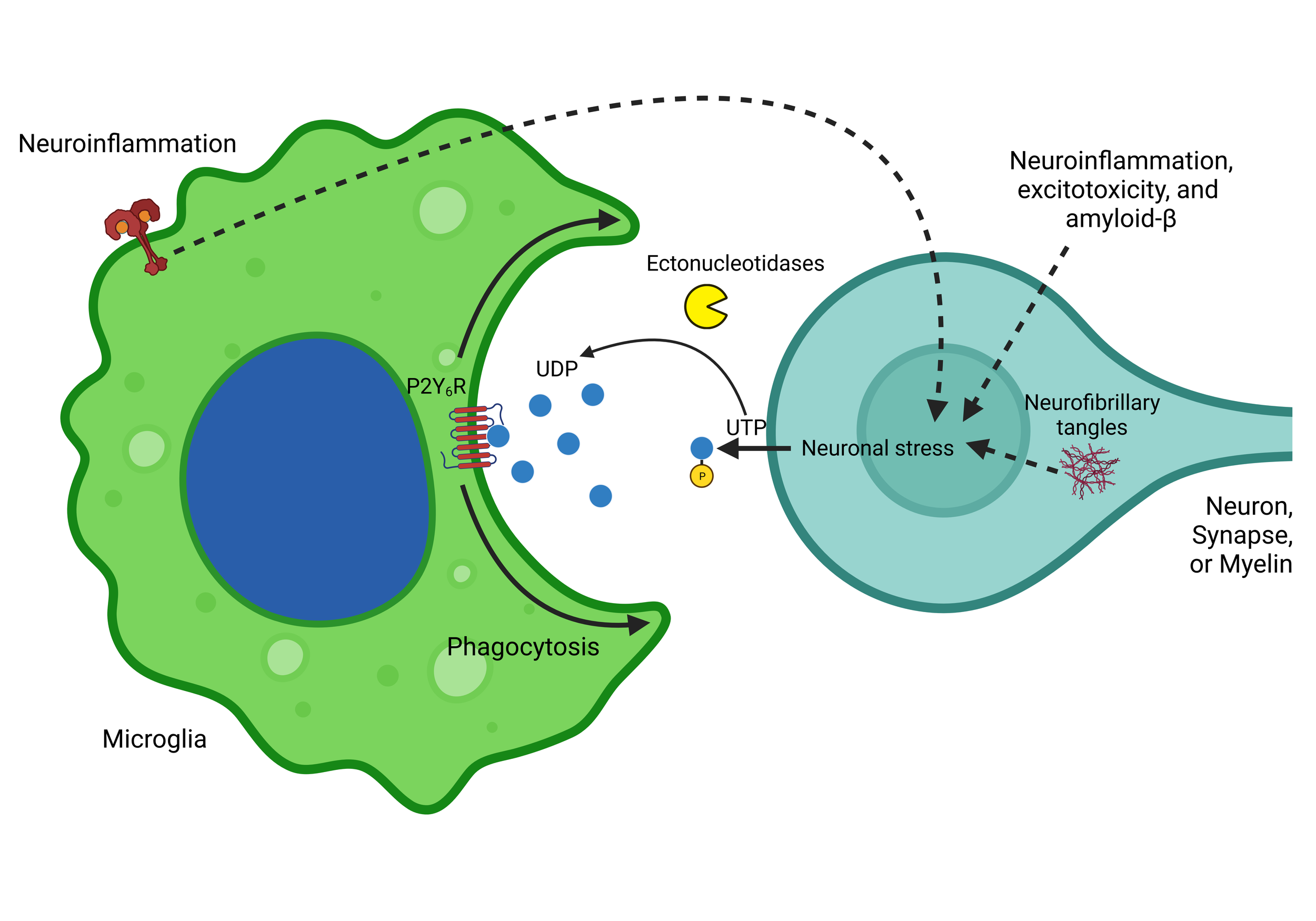
Abstract
Neurodegenerative diseases are associated with chronic neuroinflammation in the brain, which can result in microglial phagocytosis of live synapses and neurons that may contribute to cognitive deficits and neuronal loss. The microglial P2Y6 receptor (P2Y6R) is a G-protein coupled receptor, which stimulates microglial phagocytosis when activated by extracellular uridine diphosphate, released by stressed neurons. Knockout or inhibition of P2Y6R can prevent neuronal loss in mouse models of Alzheimer's disease (AD), Parkinson's disease, epilepsy, neuroinflammation and aging, and prevent cognitive deficits in models of AD, epilepsy and aging. This review summarises the known roles of P2Y6R in the physiology and pathology of the brain, and its potential as a therapeutic target to prevent neurodegeneration and other brain pathologies.
Dundee JM, Brown GC (2024). The microglial P2Y6 receptor as a therapeutic target for neurodegenerative diseases. Transl Neurodegener. 13:47.
Bioenergetic myths of energy transduction in eukaryotic cells
News Summary:
Bioenergetics is the scientific study of energy in biology. However, Bioenergetics has confined itself to studying ATP production, based on the mistaken assumptions that ATP production = energy production (mainly by mitochondria), and that ATP consumption = energy use. Whereas, in fact, no biological processes produces energy, and energy transduction occurs throughout the cell.

Abstract
The study of energy transduction in eukaryotic cells has been divided between Bioenergetics and Physiology, reflecting and contributing to a variety of Bioenergetic myths considered here: 1) ATP production = energy production, 2) energy transduction is confined to mitochondria (plus glycolysis and chloroplasts), 3) mitochondria only produce heat when required, 4) glycolysis is inefficient compared to mitochondria, and 5) mitochondria are the main source of reactive oxygen species (ROS) in cells. These myths constitute a 'mitocentric' view of the cell that is wrong or unbalanced. In reality, mitochondria are the main site of energy dissipation and heat production in cells, and this is an essential function of mitochondria in mammals. Energy transduction and ROS production occur throughout the cell, particularly the cytosol and plasma membrane, and all cell membranes act as two-dimensional energy conduits. Glycolysis is efficient, and produces less heat per ATP than mitochondria, which might explain its increased use in muscle and cancer cells.
Brown GC. Bioenergetic myths of energy transduction in eukaryotic cells. Front Mol Biosci. 2024 Jun 17;11:1402910.
Stopping the aged brain from eating itself
News summary:
The brain shrinks with age, accompanied by a loss of synapses and memory. We outline here recent evidence in mice that this loss is due to microglial phagocytosis of the synapses, mediated by the microglial P2Y6 receptor (P2Y6R).
Puigdellívol M, Brown GC. Stopping the aged brain from eating itself. Aging (Albany NY). 2024 May 7;16(9):7508-7510.
Abstract
Brain atrophy during aging appears to be partly due to brain cells, called microglia, eating bits of neurons and the connections between neurons, called synapses. Brain shrinkage and loss of synapses correlate with age-associated memory impairment, which affects 50% of people over 60 years old, causing reduced well-being, mental function, and economic activity. There is evidence in mice that aging-induced loss of synapses and memory is due to the phagocytosis (i.e. eating) of synapses by microglia. Microglial phagocytosis is regulated by several factors, including the microglial P2Y6 receptor (P2Y6R) activated by extracellular UDP (uridine diphosphate). We recently reported that microglial phagocytosis of synapses during aging is mediated by P2Y6R. Inhibition or knockout of P2Y6R reduced microglial phagocytosis of synapses and synaptic loss in co-cultures of neurons and microglia. In vivo, microglial phagocytosis of synapses was increased in the brains of aged (17 months old) wild-type mice, compared to adult (4 months old) mice, but this increase was absent in P2Y6R knockout mice. P2Y6R knockout mice also had reduced aging-associated loss of synapses and memory. Thus, inhibiting P2Y6R can reduce the loss of synapses and memory with age in mice, probably by preventing microglial phagocytosis of synapses.
News summary:
Alzheimer’s disease patients have increased levels of lipopolysaccharide (LPS) in their blood. We outline here: the hypothesis that LPS is a contributory cause of this disease, the evidence for this hypothesis, and potential treatments based on the hypothesis.
Brown GC, Heneka MT (2024) The endotoxin hypothesis of Alzheimer's disease. Mol Neurodegener. 19:30.
Abstract
Lipopolysaccharide (LPS) constitutes much of the surface of Gram-negative bacteria, and if LPS enters the human body or brain can induce inflammation and act as an endotoxin. We outline the hypothesis here that LPS may contribute to the pathophysiology of Alzheimer's disease (AD) via peripheral infections or gut dysfunction elevating LPS levels in blood and brain, which promotes: amyloid pathology, tau pathology and microglial activation, contributing to the neurodegeneration of AD. The evidence supporting this hypothesis includes: i) blood and brain levels of LPS are elevated in AD patients, ii) AD risk factors increase LPS levels or response, iii) LPS induces Aβ expression, aggregation, inflammation and neurotoxicity, iv) LPS induces TAU phosphorylation, aggregation and spreading, v) LPS induces microglial priming, activation and neurotoxicity, and vi) blood LPS induces loss of synapses, neurons and memory in AD mouse models, and cognitive dysfunction in humans. However, to test the hypothesis, it is necessary to test whether reducing blood LPS reduces AD risk or progression. If the LPS endotoxin hypothesis is correct, then treatments might include: reducing infections, changing gut microbiome, reducing leaky gut, decreasing blood LPS, or blocking LPS response.
News summary: We developed a new way to screen for drugs and drug targets that prevent inflammation-induced loss of neurons. Co-cultures of brain neurons, astrocytes and microglia were exposed to inflammation ± 227 compounds known to target particular cellular proteins, and 3 days later the cultures were analysed using image analysis software. We found a surprising set of compounds and pathways protected against inflammation-induced neuronal loss. This works may aid both drug development and understanding of brain pathology.
Abstract
Neuropathology is often mediated by interactions between neurons and glia that cannot be modeled by monocultures. However, cocultures are difficult to use and analyze for high-content screening. Here, we perform compound screening using primary neuron-glia cultures to model inflammatory neurodegeneration, live-cell stains, and automated classification of neurons, astrocytes or microglia using open-source software. Out of 227 compounds with known bioactivities, 29 protected against lipopolysaccharide-induced neuronal loss, including drugs affecting adrenergic, steroid, inflammatory and MAP kinase signaling. The screen also identified physiological compounds, such as noradrenaline and progesterone, that protected and identified neurotoxic compounds, such as a TLR7 agonist, that induced microglial proliferation. Most compounds used here have not been tested in a neuron-glia coculture neurodegeneration assay previously. Thus, combining a complex cellular disease model with high-content screening of known compounds and automated image analysis allows identification of important biology, as well as potential targets and drugs for treatment.
Birkle TJY, Willems HMG, Skidmore J, Brown GC (2024). Disease phenotypic screening in neuron-glia cocultures identifies blockers of inflammatory neurodegeneration. iScience. 27:109454.
Activated microglia release β−galactosidase that promotes inflammatory neurodegeneration
News Summary: We find that inflamed and aged microglia release an enzyme β-galactosidase that activates microglia to cause neuronal loss, and this can be prevented by inhibiting β-galactosidase. Thus, inhibiting this released enzyme might be beneficial in neurodegeneration or aging.
Kitchener EJA, Dundee JM and Brown GC (2024) . Front. Aging Neurosci. 15:1327756.
Abstract
Beta (β)-galactosidase is a lysosomal enzyme that removes terminal galactose residues from glycolipids and glycoproteins. It is upregulated in, and used as a marker for, senescent cells. Microglia are brain macrophages implicated in neurodegeneration, and can upregulate β-galactosidase when senescent. We find that inflammatory activation of microglia induced by lipopolysaccharide results in translocation of β-galactosidase to the cell surface and release into the medium. Similarly, microglia in aged mouse brains appear to have more β-galactosidase on their surface. Addition of β-galactosidase to neuronal-glial cultures causes microglial activation and neuronal loss mediated by microglia. Inhibition of β-galactosidase in neuronal-glial cultures reduces inflammation and neuronal loss induced by lipopolysaccharide. Thus, activated microglia release β-galactosidase that promotes microglial-mediated neurodegeneration which is prevented by inhibition of β-galactosidase.
LRPAP1 is released from activated microglia and inhibits microglial phagocytosis and amyloid beta aggregation
News summary: RAP (also known as LRPAP1) is a chaperone for LDL family receptors, including LRP1, in the endoplasmic reticulum (ER). Extracellular RAP can also potently inhibit these receptors. We find that RAP is released by inflamed or stressed microglia, resulting in nanomolar levels of extracellular RAP. And these levels of RAP strongly inhibited microglial phagocytosis. and amyloid beta aggregation. Thus, RAP appears to be a potent regulator of microglia and amyloid.
Reid KM, Brown GC (2023) LRPAP1 is released from activated microglia and inhibits microglial phagocytosis and amyloid beta aggregation. Front Immunol. 14:1286474.
Abstract
Low-density lipoprotein receptor-related protein-associated protein 1 (LRPAP1), also known as receptor associated protein (RAP), is an endoplasmic reticulum (ER) chaperone and inhibitor of LDL receptor related protein 1 (LRP1) and related receptors. These receptors have dozens of physiological ligands and cell functions, but it is not known whether cells release LRPAP1 physiologically at levels that regulate these receptors and cell functions. We used mouse BV-2 and human CHME3 microglial cell lines, and found that microglia released nanomolar levels of LRPAP1 when inflammatory activated by lipopolysaccharide or when ER stressed by tunicamycin. LRPAP1 was found on the surface of live activated and non-activated microglia, and anti-LRPAP1 antibodies induced internalization. Addition of 10 nM LRPAP1 inhibited microglial phagocytosis of isolated synapses and cells, and the uptake of Aβ. LRPAP1 also inhibited Aβ aggregation in vitro. Thus, activated and stressed microglia release LRPAP1 levels that can inhibit phagocytosis, Aβ uptake and Aβ aggregation. We conclude that LRPAP1 release may regulate microglial functions and Aβ pathology, and more generally that extracellular LRPAP1 may be a physiological and pathological regulator of a wide range of cell functions.
P2Y6 receptor-dependent microglial phagocytosis of synapses during development regulates synapse density and memory
News summary: The P2Y6 receptor (P2Y6R) is activated by UDP released by neurons, inducing microglial phagocytosis of such neurons or synapses. We tested whether P2Y6R regulates developmental synaptic pruning in mice and found that P2Y6R knockout mice have reduced synaptic material within microglial lysosomes, and increased synaptic density in the brains of postnatal day 30 mice, consistent with reduced synaptic pruning during development. We also found that adult P2Y6R knockout mice had reduced memory, consistent with persistent deficits in brain function, resulting from impaired synaptic pruning. Overall, the results suggest that P2Y6R mediates microglial phagocytosis of synapses during development, and the absence of this results in memory deficits in the adult.
Dundee JM, Puigdellívol M, Butler R, Brown GC. P2Y6 receptor-dependent microglial phagocytosis of synapses during development regulates synapse density and memory. J Neurosci. 2023 Sep 27:JN-RM-1089-23.
https://doi.org/10.1523/JNEUROSCI.1089-23.2023
Abstract
During brain development, excess synapses are pruned (i.e. removed), in part by microglial phagocytosis, and dysregulated synaptic pruning can lead to behavioral deficits. The P2Y6 receptor (P2Y6R) is known to regulate microglial phagocytosis of neurons, and to regulate microglial phagocytosis of synapses in cell culture and in vivo during aging. However, currently it is unknown whether P2Y6R regulates synaptic pruning during development. Here, we show that P2Y6R knockout mice had strongly reduced microglial internalization of synaptic material, measured as Vglut1 within CD68-staining lysosomes of microglia at postnatal day 30 (P30), suggesting reduced microglial phagocytosis of synapses. Consistent with this, we found an increased density of synapses in the somatosensory cortex and the CA3 region and dentate gyrus of the hippocampus at P30. We also show that adult P2Y6R knockout mice have impaired short- and long-term spatial memory and impaired short- and long-term recognition memory compared to wild-type mice, as measured by novel location recognition, novel object recognition, and Y-maze memory tests. Overall, this indicates P2Y6R regulates microglial phagocytosis of synapses during development, and this contributes to memory capacity.
Cell death by phagocytosis
News summary: One of the oldest known ways in which a cell can die is as a result of the cell being eaten alive by another cell, and then being killed and digested within the engulfing cell. This is called ‘cell death by phagocytosis’, or ‘phagoptosis’ for short. Recent research has shown that this form of cell death is very important during mammalian development, tissue turnover, immunity, cancer, neurology and ageing.
Brown GC. Cell death by phagocytosis. Nat Rev Immunol. 2023 Aug 21.
https://doi.org/10.1038/s41577-023-00921-6
Abstract
Cells can die as a consequence of being phagocytosed by other cells - a form of cell death that has been called phagotrophy, cell cannibalism, programmed cell removal and primary phagocytosis. However, these are all different manifestations of cell death by phagocytosis (termed 'phagoptosis' for short). The engulfed cells die as a result of cytotoxic oxidants, peptides and degradative enzymes within acidic phagolysosomes. Cell death by phagocytosis was discovered by Metchnikov in the 1880s, but was neglected until recently. It is now known to contribute to developmental cell death in nematodes, Drosophila and mammals, and is central to innate and adaptive immunity against pathogens. Cell death by phagocytosis mediates physiological turnover of erythrocytes and other leucocytes, making it the most abundant form of cell death in the mammalian body. Immunity against cancer is also partly mediated by macrophage phagocytosis of cancer cells, but cancer cells can also phagocytose host cells and other cancer cells in order to survive. Recent evidence indicates neurodegeneration and other neuropathologies can be mediated by microglial phagocytosis of stressed neurons. Thus, despite cell death by phagocytosis being poorly recognized, it is one of the oldest, commonest and most important forms of cell death.
Extracellular tau stimulates phagocytosis of living neurons by activated microglia via Toll-like 4 receptor-NLRP3 inflammasome-caspase-1 signalling axis
News summary: Alzheimer’s disease involves the aggregation and spreading of a protein called tau. We find here that extracellular tau activates microglia via Toll-like receptor 4 (TLR4), inducing the inflammasome, resulting in caspase-1 activation, which stimulates microglial phagocytosis. Meanwhile, oxidants from the NADPH oxidase cause neurons to expose phosphatidyserine, which induces microglia to phagocytose these live neurons, resulting in neuronal loss. This research suggests multiple ways to prevent neuronal loss during the neurodegeneration induced by tau.
Pampuscenko K, Morkuniene R, Krasauskas L, Smirnovas V, Brown GC, Borutaite V. Extracellular tau stimulates phagocytosis of living neurons by activated microglia via Toll-like 4 receptor-NLRP3 inflammasome-caspase-1 signalling axis. Sci Rep. 2023 Jul 4;13(1):10813.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10319744/
Abstract
In tauopathies, abnormal deposition of intracellular tau protein followed by gradual elevation of tau in cerebrospinal fluids and neuronal loss has been documented, however, the mechanism how actually neurons die under tau pathology is largely unknown. We have previously shown that extracellular tau protein (2N4R isoform) can stimulate microglia to phagocytose live neurons, i.e. cause neuronal death by primary phagocytosis, also known as phagoptosis. Here we show that tau protein induced caspase-1 activation in microglial cells via 'Toll-like' 4 (TLR4) receptors and neutral sphingomyelinase. Tau-induced neuronal loss was blocked by caspase-1 inhibitors (Ac-YVAD-CHO and VX-765) as well as by TLR4 antibodies. Inhibition of caspase-1 by Ac-YVAD-CHO prevented tau-induced exposure of phosphatidylserine on the outer leaflet of neuronal membranes and reduced microglial phagocytic activity. We also show that suppression of NLRP3 inflammasome, which is down-stream of TLR4 receptors and mediates caspase-1 activation, by a specific inhibitor (MCC550) also prevented tau-induced neuronal loss. Moreover, NADPH oxidase is also involved in tau-induced neurotoxicity since neuronal loss was abolished by its pharmacological inhibitor. Overall, our data indicate that extracellular tau protein stimulates microglia to phagocytose live neurons via Toll-like 4 receptor-NLRP3 inflammasome-caspase-1 axis and NADPH oxidase, each of which may serve as a potential molecular target for pharmacological treatment of tauopathies.
The Endotoxin Hypothesis of Parkinson's Disease
News summary: We propose and review evidence for the hypothesis that Parkinson’s disease is in part caused by the endotoxin lipopolysaccharide. In early Parkinson’s disease, endotoxin from Gram-negative bacteria in the gut leaks into and across the gut wall causing inflammation, elevated blood endotoxin and aggregation of alpha synuclein, which together induce neuronal loss in the brain.
Brown GC, Camacho M, Williams-Gray CH. The Endotoxin Hypothesis of Parkinson's Disease. Mov Disord. 2023 May 8.
Abstract
The endotoxin hypothesis of Parkinson's disease (PD) is the idea that lipopolysaccharide (LPS) endotoxins contribute to the pathogenesis of this disorder. LPS endotoxins are found in, and released from, the outer membrane of Gram-negative bacteria, for example in the gut. It is proposed that gut dysfunction in early PD leads to elevated LPS levels in the gut wall and blood, which promotes both α-synuclein aggregation in the enteric neurons and a peripheral inflammatory response. Communication to the brain via circulating LPS and cytokines in the blood and/or the gut-brain axis leads to neuroinflammation and spreading of α-synuclein pathology, exacerbating neurodegeneration in brainstem nuclei and loss of dopaminergic neurons in the substantia nigra, and manifesting in the clinical symptoms of PD. The evidence supporting this hypothesis includes: (1) gut dysfunction, permeability, and bacterial changes occur early in PD, (2) serum levels of LPS are increased in a proportion of PD patients, (3) LPS induces α-synuclein expression, aggregation, and neurotoxicity, (4) LPS causes activation of peripheral monocytes leading to inflammatory cytokine production, and (5) blood LPS causes brain inflammation and specific loss of midbrain dopaminergic neurons, mediated by microglia. If the hypothesis is correct, then treatment options might include: (1) changing the gut microbiome, (2) reducing gut permeability, (3) reducing circulating LPS levels, or (4) blocking the response of immune cells and microglia to LPS. However, the hypothesis has a number of limitations and requires further testing, in particular whether reducing LPS levels can reduce PD incidence, progression, or severity.
Syk inhibitors protect against microglia-mediated neuronal loss in culture
News summary
Spleen tyrosine kinase (Syk) is activated by multiple microglial receptors implicated in neurodegeneration. So we tested whether Syk inhibitors could prevent neuronal loss in brain cell cultures. We found that two different Syk inhibitors could fully prevent neuronal loss induced by lipopolysaccharide (LPS), as well as spontaneous neuronal loss. The mechanism of this protection appears to be via Syk inhibitors blocking microglial phagocytosis, cytokine release and survival. As these Syk inhibitors have been used clinically for cancer, they might be useful also to prevent neurodegeneration.
Birkle TJY & Brown GC (2023) Syk inhibitors protect against microglia-mediated neuronal loss in culture. Front. Aging Neurosci., 15 March 2023
Abstract
Microglia are brain macrophages and play beneficial and/or detrimental roles in many brain pathologies because of their inflammatory and phagocytic activity. Microglial inflammation and phagocytosis are thought to be regulated by spleen tyrosine kinase (Syk), which is activated by multiple microglial receptors, including TREM2 (Triggering Receptor Expressed on Myeloid Cells 2), implicated in neurodegeneration. Here, we have tested whether Syk inhibitors can prevent microglia-dependent neurodegeneration induced by lipopolysaccharide (LPS) in primary neuron-glia cultures. We found that the Syk inhibitors BAY61-3606 and P505-15 (at 1 and 10 μM, respectively) completely prevented the neuronal loss induced by LPS, which was microglia-dependent. Syk inhibition also prevented the spontaneous loss of neurons from older neuron-glia cultures. In the absence of LPS, Syk inhibition depleted microglia from the cultures and induced some microglial death. However, in the presence of LPS, Syk inhibition had relatively little effect on microglial density (reduced by 0–30%) and opposing effects on the release of two pro-inflammatory cytokines (IL-6 decreased by about 45%, TNFα increased by 80%). Syk inhibition also had no effect on the morphological transition of microglia exposed to LPS. On the other hand, inhibition of Syk reduced microglial phagocytosis of beads, synapses and neurons. Thus, Syk inhibition in this model is most likely neuroprotective by reducing microglial phagocytosis, however, the reduced microglial density and IL-6 release may also contribute. This work adds to increasing evidence that Syk is a key regulator of the microglial contribution to neurodegenerative disease and suggests that Syk inhibitors may be used to prevent excessive microglial phagocytosis of synapses and neurons.
P2Y6 receptor-dependent microglial phagocytosis of synapses mediates synaptic and memory loss in aging
News summary
We find that the P2Y6 receptor on microglia regulates microglial phagocytosis of synapses in culture. In vivo, P2Y6 receptor knockout in mice prevents: an increase in microglial phagocytosis of synapses, a decrease in synaptic density and a loss of memory, induced by aging. Thus, blocking the P2Y6 receptor prevents aging-induced memory loss, apparently by preventing microglial phagocytosis of synapses.
Dundee JM, Puigdellívol M, Butler R, Cockram TOJ, Brown GC (2023) P2Y6 receptor-dependent microglial phagocytosis of synapses mediates synaptic and memory loss in aging. Aging Cell 22:e13761.
Abstract
Aging causes loss of brain synapses and memory, and microglial phagocytosis of synapses may contribute to this loss. Stressed neurons can release the nucleotide UTP, which is rapidly converted into UDP, that in turn activates the P2Y6 receptor (P2Y6 R) on the surface of microglia, inducing microglial phagocytosis of neurons. However, whether the activation of P2Y6 R affects microglial phagocytosis of synapses is unknown. We show here that inactivation of P2Y6 R decreases microglial phagocytosis of isolated synapses (synaptosomes) and synaptic loss in neuronal-glial co-cultures. In vivo, wild-type mice aged from 4 to 17 months exhibited reduced synaptic density in cortical and hippocampal regions, which correlated with increased internalization of synaptic material within microglia. However, this aging-induced synaptic loss and internalization were absent in P2Y6 R knockout mice, and these mice also lacked any aging-induced memory loss. Thus, P2Y6 R appears to mediate aging-induced loss of synapses and memory by increasing microglial phagocytosis of synapses. Consequently, blocking P2Y6 R has the potential to prevent age-associated memory impairment.
Alzheimer's disease-associated R47H TREM2 increases, but wild-type TREM2 decreases, microglial phagocytosis of synaptosomes and neuronal loss
News summary: TREM2 is a receptor regulating microglial phagocytosis. A single copy of the R47H variant of the TREM2 gene increases the risk of developing Alzheimer’s disease about four fold, but the mechanism is unclear. We expressed R47H or common variant TREM2 in different microglial cells. Surprisingly, the common variant of TREM2 inhibited phagocytosis via inducing the expression of cystatin F. Whereas R47H TREM2 increased phagocytosis of synapse and neurons, apparently because R47H TREM2 has increased activation by phosphatidylserine. These data suggest that R47H TREM2 may increase AD risk by increasing phagocytosis of synapses and neurons via greater activation by phosphatidylserine and that WT TREM2 may decrease microglial phagocytosis of synapses and neurons via cystatin F.
Popescu AS, Butler CA, Allendorf DH, Piers TM, Mallach A, Roewe J, Reinhardt P, Cinti A, Redaelli L, Boudesco C, Pradier L, Pocock JM, Thornton P, Brown GC (2023) Alzheimer's disease-associated R47H TREM2 increases, but wild-type TREM2 decreases, microglial phagocytosis of synaptosomes and neuronal loss. Glia 71:974-990.
DOI: 10.1002/glia.24318
Abstract
Triggering receptor on myeloid cells 2 (TREM2) is an innate immune receptor, upregulated on the surface of microglia associated with amyloid plaques in Alzheimer's disease (AD). Individuals heterozygous for the R47H variant of TREM2 have greatly increased risk of developing AD. We examined the effects of wild-type (WT), R47H and knock-out (KO) of human TREM2 expression in three microglial cell systems. Addition of mouse BV-2 microglia expressing R47H TREM2 to primary mouse neuronal cultures caused neuronal loss, not observed with WT TREM2. Neuronal loss was prevented by using annexin V to block exposed phosphatidylserine, an eat-me signal and ligand of TREM2, suggesting loss was mediated by microglial phagocytosis of neurons exposing phosphatidylserine. Addition of human CHME-3 microglia expressing R47H TREM2 to LUHMES neuronal-like cells also caused loss compared to WT TREM2. Expression of R47H TREM2 in BV-2 and CHME-3 microglia increased their uptake of phosphatidylserine-beads and synaptosomes versus WT TREM2. Human iPSC-derived microglia with heterozygous R47H TREM2 had increased phagocytosis of synaptosomes vs common-variant TREM2. Additionally, phosphatidylserine liposomes increased activation of human iPSC-derived microglia expressing homozygous R47H TREM2 versus common-variant TREM2. Finally, overexpression of TREM2 in CHME-3 microglia caused increased expression of cystatin F, a cysteine protease inhibitor, and knock-down of cystatin F increased CHME-3 uptake of phosphatidylserine-beads. Together, these data suggest that R47H TREM2 may increase AD risk by increasing phagocytosis of synapses and neurons via greater activation by phosphatidylserine and that WT TREM2 may decrease microglial phagocytosis of synapses and neurons via cystatin F.
Neu1 is released from activated microglia, stimulating microglial phagocytosis and sensitizing neurons to glutamate
News summary: Neuraminidase 1 (Neu1) is a sialidase, i.e. an enzyme that can remove terminal sialic acid residues from glycoproteins and glycoproteins. Neu1 is normally in lysosomes, but we found LPS-activated microglia released Neu1 into the medium, probably by lysosomal exocytosis. Extracellular Neu1 stimulated microglial phagocytosis and sensitised neurons to glutamate. This indicates that activated microglia can change brain functions via altering cell surface sialylation of both microglia and neurons.
Allendorf DH and Brown GC (2022) Neu1 Is Released From Activated Microglia, Stimulating Microglial Phagocytosis and Sensitizing Neurons to Glutamate. Front. Cell. Neurosci. 16:917884.
https:/doi.org/10.3389/fncel.2022.917884
Abstract
Neuraminidase 1 (Neu1) hydrolyses terminal sialic acid residues from glycoproteins and glycolipids, and is normally located in lysosomes, but can be released onto the surface of activated myeloid cells and microglia. We report that endotoxin/lipopolysaccharide-activated microglia released Neu1 into culture medium, and knockdown of Neu1 in microglia reduced both Neu1 protein and neuraminidase activity in the culture medium. Release of Neu1 was reduced by inhibitors of lysosomal exocytosis, and accompanied by other lysosomal proteins, including protective protein/cathepsin A, known to keep Neu1 active. Extracellular neuraminidase or over-expression of Neu1 increased microglial phagocytosis, while knockdown of Neu1 decreased phagocytosis. Microglial activation caused desialylation of microglial phagocytic receptors Trem2 and MerTK, and increased binding to Trem2 ligand galectin-3. Culture media from activated microglia contained Neu1, and when incubated with neurons induced their desialylation, and increased the neuronal death induced by low levels of glutamate. Direct desialylation of neurons by adding sialidase or inhibiting sialyltransferases also increased glutamate-induced neuronal death. We conclude that activated microglia can release active Neu1, possibly by lysosomal exocytosis, and this can both increase microglial phagocytosis and sensitize neurons to glutamate, thus potentiating neuronal death.
Microglia states and nomenclature: A field at its crossroads
Summary
Microglial research has advanced considerably in recent decades yet has been constrained by a rolling series of dichotomies such as "resting versus activated" and "M1 versus M2." This dualistic classification of good or bad microglia is inconsistent with the wide repertoire of microglial states and functions in development, plasticity, aging, and diseases that were elucidated in recent years. New designations continuously arising in an attempt to describe the different microglial states, notably defined using transcriptomics and proteomics, may easily lead to a misleading, although unintentional, coupling of categories and functions. To address these issues, we assembled a group of multidisciplinary experts to discuss our current understanding of microglial states as a dynamic concept and the importance of addressing microglial function. Here, we provide a conceptual framework and recommendations on the use of microglial nomenclature for researchers, reviewers, and editors, which will serve as the foundations for a future white paper.
Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, Amit I, Audinat E, Bechmann I, Bennett M, Bennett F, Bessis A, Biber K, Bilbo S, Blurton-Jones M, Boddeke E, Brites D, Brône B, Brown GC, Butovsky O, Carson MJ, Castellano B, Colonna M, Cowley SA, Cunningham C, Davalos D, De Jager PL, de Strooper B, Denes A, Eggen BJL, Eyo U, Galea E, Garel S, Ginhoux F, Glass CK, Gokce O, Gomez-Nicola D, González B, Gordon S, Graeber MB, Greenhalgh AD, Gressens P, Greter M, Gutmann DH, Haass C, Heneka MT, Heppner FL, Hong S, Hume DA, Jung S, Kettenmann H, Kipnis J, Koyama R, Lemke G, Lynch M, Majewska A, Malcangio M, Malm T, Mancuso R, Masuda T, Matteoli M, McColl BW, Miron VE, Molofsky AV, Monje M, Mracsko E, Nadjar A, Neher JJ, Neniskyte U, Neumann H, Noda M, Peng B, Peri F, Perry VH, Popovich PG, Pridans C, Priller J, Prinz M, Ragozzino D, Ransohoff RM, Salter MW, Schaefer A, Schafer DP, Schwartz M, Simons M, Smith CJ, Streit WJ, Tay TL, Tsai LH, Verkhratsky A, von Bernhardi R, Wake H, Wittamer V, Wolf SA, Wu LJ, Wyss-Coray T. Microglia states and nomenclature: A field at its crossroads. Neuron. 2022 Nov 2;110(21):3458-3483.
Brain Cells Release Calreticulin That Attracts and Activates Microglia, and Inhibits Amyloid Beta Aggregation and Neurotoxicity
News summary: We find that when microglial cells are inflamed, stressed or killed, they release the protein calreticulin into the extracellular space. And this extracellular calreticulin can recruit and activate other microglia. Extracellular calreticulin can also block fibrillisation of amyloid beta, and prevent the neuronal loss induced by amyloid beta. Thus, when microglia or neurons release calreticulin, it protects the brain by recruiting microglia, blocking aggregation of extracellular proteins and preventing neuronal loss.
Reid K.M., Kitchener E.J. A., Butler C.A., Cockram T O. J., Brown G.C. Brain Cells Release Calreticulin That Attracts and Activates Microglia, and Inhibits Amyloid Beta Aggregation and Neurotoxicity. Front. Immunol. 13, April 2022
https://doi.org/10.3389/fimmu.2022.859686
Abstract
Calreticulin is a chaperone, normally found in the endoplasmic reticulum, but can be released by macrophages into the extracellular medium. It is also found in cerebrospinal fluid bound to amyloid beta (Aβ). We investigated whether brain cells release calreticulin, and whether extracellular calreticulin had any effects on microglia and neurons relevant to neuroinflammation and neurodegeneration. We found that microglia release nanomolar levels of calreticulin when inflammatory-activated with lipopolysaccharide, when endoplasmic reticulum stress was induced by tunicamycin, or when cell death was induced by staurosporine, and that neurons release calreticulin when crushed. Addition of nanomolar levels of extracellular calreticulin was found to chemoattract microglia, and activate microglia to release cytokines TNF-α, IL-6 and IL-1β, as well as chemokine (C-C motif) ligand 2. Calreticulin blocked Aβ fibrillization and modified Aβ oligomerization, as measured by thioflavin T fluorescence and transmission electron microscopy. Extracellular calreticulin also altered microglial morphology and proliferation, and prevented Aβ-induced neuronal loss in primary neuron-glial cultures. Thus, calreticulin is released by microglia and neurons, and acts: as an alarmin to recruit and activate microglia, as an extracellular chaperone to prevent Aβ aggregation, and as a neuroprotectant against Aβ neurotoxicity.
Knockout of the P2Y6 Receptor Prevents Peri-Infarct Neuronal Loss after Transient, Focal Ischemia in Mouse Brain
News summary: Stroke causes a rapid loss of all cells in the infarct, but a delayed loss of neurons in surrounding areas of brain. This paper shows that knockout of the P2Y6 receptor, required for microglial phagocytosis of neurons, prevents the loss of neurons around the infarct. This suggests that this delayed neuronal loss is due to microglial phagocytosis of neurons (i.e. the microglia eat live neurons), and can be prevented by blocking the P2Y6 receptor.
Milde S, Brown GC. Knockout of the P2Y6 Receptor Prevents Peri-Infarct Neuronal Loss after Transient, Focal Ischemia in Mouse Brain. Int J Mol Sci. 2022 Feb 19;23(4):2304.
doi: 10.1016/j.celrep.2021.110148
Abstract
After stroke, there is a delayed neuronal loss in brain areas surrounding the infarct, which may in part be mediated by microglial phagocytosis of stressed neurons. Microglial phagocytosis of stressed or damaged neurons can be mediated by UDP released from stressed neurons activating the P2Y6 receptor on microglia, inducing microglial phagocytosis of such neurons. We show evidence here from a small trial that the knockout of the P2Y6 receptor, required for microglial phagocytosis of neurons, prevents the delayed neuronal loss after transient, focal brain ischemia induced by endothelin-1 injection in mice. Wild-type mice had neuronal loss and neuronal nuclear material within microglia in peri-infarct areas. P2Y6 receptor knockout mice had no significant neuronal loss in peri-infarct brain areas seven days after brain ischemia. Thus, delayed neuronal loss after stroke may in part be mediated by microglial phagocytosis of stressed neurons, and the P2Y6 receptor is a potential treatment target to prevent peri-infarct neuronal loss.
Does Soluble TREM2 Protect Against Alzheimer's Disease?
News summary: TREM2 can be cleaved extracellularly to release soluble TREM2 (sTREM2). There is evidence that people with higher levels of sTREM2 in their brains have slower progression into and through Alzheimer’s disease (AD). And there is evidence in vitro and in animals that sTREM2 is protective by: i) binding to and blocking the effects of amyloid beta, and ii) activating microglia. Thus, sTREM2 appears to protect against AD.
Brown GC, St George-Hyslop P. Does Soluble TREM2 Protect Against Alzheimer's Disease? Front Aging Neurosci. 2022 Jan 28;13:834697.
doi: 10.3389/fnagi.2021.834697.
Abstract
Triggering Receptor Expressed in Myeloid Cells 2 (TREM2) is a pattern recognition receptor on myeloid cells, and is upregulated on microglia surrounding amyloid plaques in Alzheimer's disease (AD). Rare, heterozygous mutations in TREM2 (e.g., R47H) increase AD risk several fold. TREM2 can be cleaved at the plasma membrane by metalloproteases to release the ectodomain as soluble TREM2 (sTREM2). Wild-type sTREM2 binds oligomeric amyloid beta (Aβ) and acts as an extracellular chaperone, blocking and reversing Aβ oligomerization and fibrillization, and preventing Aβ-induced neuronal loss in vitro. Whereas, R47H sTREM2 increases Aβ fibrillization and neurotoxicity. AD brains expressing R47H TREM2 have more fibrous plaques with more neuritic pathology around these plaques, consistent with R47H sTREM2 promoting Aβ fibrillization relative to WT sTREM2. Brain expression or injection of wild-type sTREM2 reduces pathology in amyloid models of AD in mice, indicating that wild-type sTREM2 is protective against amyloid pathology. Levels of sTREM2 in cerebrospinal fluid (CSF) fall prior to AD, rise in early AD, and fall again in late AD. People with higher sTREM2 levels in CSF progress more slowly into and through AD than do people with lower sTREM2 levels, suggesting that sTREM2 protects against AD. However, some of these experiments can be interpreted as full-length TREM2 protecting rather than sTREM2, and to distinguish between these two possibilities, we need more experiments testing whether sTREM2 itself protects in AD and AD models, and at what stage of disease. If sTREM2 is protective, then treatments could be designed to elevate sTREM2 in AD.
The microglial P2Y6 receptor mediates neuronal loss and memory deficits in neurodegeneration
News summary: The mechanisms by which neurons are lost in neurodegenerative diseases are unclear. This paper shows that knockout of the P2Y6 receptor, required for microglial phagocytosis of neurons, prevents the microglial phagocytosis of neurons and the loss of neurons induced by amyloid beta and tau in mouse models of neurodegenerative diseases. This suggests that this neuronal loss is due to microglial phagocytosis of neurons (i.e. the microglia eat live neurons), and can be prevented by blocking the P2Y6 receptor.
Puigdellívol M, Milde S, Vilalta A, Cockram TOJ, Allendorf DH, Lee JY, Dundee JM, Pampuščenko K, Borutaite V, Nuthall HN, Brelstaff JH, Spillantini MG, Brown GC. The microglial P2Y6 receptor mediates neuronal loss and memory deficits in neurodegeneration. Cell Rep. 2021 Dec 28;37(13):110148.
doi: 10.1016/j.celrep.2021.110148.
Abstract
Microglia are implicated in neurodegeneration, potentially by phagocytosing neurons, but it is unclear how to block the detrimental effects of microglia while preserving their beneficial roles. The microglial P2Y6 receptor (P2Y6R) - activated by extracellular UDP released by stressed neurons - is required for microglial phagocytosis of neurons. We show here that injection of amyloid beta (Aβ) into mouse brain induces microglial phagocytosis of neurons, followed by neuronal and memory loss, and this is all prevented by knockout of P2Y6R. In a chronic tau model of neurodegeneration (P301S TAU mice), P2Y6R knockout prevented TAU-induced neuronal and memory loss. In vitro, P2Y6R knockout blocked microglial phagocytosis of live but not dead targets and reduced tau-, Aβ-, and UDP-induced neuronal loss in glial-neuronal cultures. Thus, the P2Y6 receptor appears to mediate Aβ- and tau-induced neuronal and memory loss via microglial phagocytosis of neurons, suggesting that blocking this receptor may be beneficial in the treatment of neurodegenerative diseases.
Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons
News summary: This paper reviews the evidence that the neuronal loss that occurs some days after stroke is due to microglia phagocytosing (eating) stressed neurons. These neurons release or bind signals that encourage microglia to phagocytose the stressed neurons, and therefore this neuronal loss can be prevented by inhibiting this microglial phagocytosis.
Brown, G.C. 2021. Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons. Int. J. Mol. Sci., 22(24).
Abstract
After stroke, there is a rapid necrosis of all cells in the infarct, followed by a delayed loss of neurons both in brain areas surrounding the infarct, known as ‘selective neuronal loss’, and in brain areas remote from, but connected to, the infarct, known as ‘secondary neurodegeneration’. Here we review evidence indicating that this delayed loss of neurons after stroke is mediated by the microglial phagocytosis of stressed neurons. After a stroke, neurons are stressed by ongoing ischemia, excitotoxicity and/or inflammation and are known to: (i) release “find-me” signals such as ATP, (ii) expose “eat-me” signals such as phosphatidylserine, and (iii) bind to opsonins, such as complement components C1q and C3b, inducing microglia to phagocytose such neurons. Blocking these factors on neurons, or their phagocytic receptors on microglia, can prevent delayed neuronal loss and behavioral deficits in rodent models of ischemic stroke. Phagocytic receptors on microglia may be attractive treatment targets to prevent delayed neuronal loss after stroke due to the microglial phagocytosis of stressed neurons.
Inflammatory neuronal loss in the substantia nigra induced by systemic lipopolysaccharide is prevented by knockout of the P2Y6 receptor in mice
News summary: Inflammation in the body induced by endotoxin injections in mice causes neuronal loss in the brain, similar to that in Parkinson’s disease, and can be prevented by blocking a microglial receptor. This supports the hypotheses that Parkinson’s is caused by blood endotoxin, and can be treated by blocking the P2Y6 receptor.
Milde, S., van Tartwijk, F. W., Vilalta, A., Hornik, T. C., Dundee, J. M., Puigdellívol, M., & Brown, G. C., 2021. Inflammatory neuronal loss in the substantia nigra induced by systemic lipopolysaccharide is prevented by knockout of the P2Y6 receptor in mice. J Neuroinflammation, 18(1).
DOI: 10.1186/s12974-021-02280-2
Abstract
Inflammation may contribute to multiple brain pathologies. One cause of inflammation is lipopolysaccharide/endotoxin (LPS), the levels of which are elevated in blood and/or brain during bacterial infections, gut dysfunction and neurodegenerative diseases, such as Parkinson's disease. How inflammation causes neuronal loss is unclear, but one potential mechanism is microglial phagocytosis of neurons, which is dependent on the microglial P2Y6 receptor. We investigated here whether the P2Y6 receptor was required for inflammatory neuronal loss. Intraperitoneal injection of LPS on 4 successive days resulted in specific loss of dopaminergic neurons (measured as cells staining with tyrosine hydroxylase or NeuN) in the substantia nigra of wild-type mice, but no neuronal loss in cortex or hippocampus. This supports the hypothesis that neuronal loss in Parkinson's disease may be driven by peripheral LPS. By contrast, there was no LPS-induced neuronal loss in P2Y6 receptor knockout mice. In vitro, LPS-induced microglial phagocytosis of cells was prevented by inhibition of the P2Y6 receptor, and LPS-induced neuronal loss was reduced in mixed glial-neuronal cultures from P2Y6 receptor knockout mice. This supports the hypothesis that microglial phagocytosis contributes to inflammatory neuronal loss, and can be prevented by blocking the P2Y6 receptor, suggesting that P2Y6 receptor antagonists might be used to prevent inflammatory neuronal loss in Parkinson's disease and other brain pathologies involving inflammatory neuronal loss.
CD33M inhibits microglial phagocytosis, migration and proliferation, but the Alzheimer’s disease-protective variant CD33m stimulates phagocytosis and proliferation, and inhibits adhesion
News summary: The microglial receptor variant, CD33m, which reduces risk of Alzheimer’s disease, causes protective microglial behaviour, despite loosing its ligand binding site. This suggest that the disease could be treated by increasing CD33m levels.
Ann Butler, C., Thornton, P. and Brown, G. C., 2021. CD33M inhibits microglial phagocytosis, migration and proliferation, but the Alzheimer’s disease‐protective variant CD33m stimulates phagocytosis and proliferation, and inhibits adhesion. Journal of Neurochemistry, 158(2).
DOI: 10.1111/jnc.15349
Abstract
CD33 is a Siglec (sialic acid-binding immunoglobulin-type lectin) receptor on microglia. Human CD33 can be alternatively spliced into two isoforms: the long isoform (CD33M) and a shorter isoform (CD33m) that lacks the sialic acid-binding site. CD33m appears to protect against Alzheimer's disease; however, it remains unclear how. To investigate potential mechanisms by which CD33m may confer protection, we expressed the CD33m and CD33M isoforms of human CD33 in mouse BV-2 and human CHME3 microglial cells and assessed microglia functions. In the BV-2 cells, CD33M inhibited microglial phagocytosis of beads, synapses, debris and dead cells, while CD33m increased phagocytosis of beads, debris and cells. RNAi knockdown of the endogenous mouse CD33 increased phagocytosis and prevented CD33m's (but not CD33M’s) effect on phagocytosis. CD33M increased cell attachment but inhibited cell proliferation, while CD33m did the opposite. We also found that CD33M inhibited cell migration. In human CHME3 cells, CD33M increased cell attachment, but inhibited phagocytosis, proliferation and migration, whereas CD33m did the opposite. We conclude that CD33M inhibits microglial phagocytosis, inhibits migration and increases adhesion, while CD33m increases phagocytosis, proliferation and inhibits adhesion. Thus, CD33m might protect against Alzheimer's disease by increasing microglial proliferation, movement and phagocytosis of debris and dead cells.
I’m Infected, Eat Me! Innate Immunity Mediated by Live, Infected Cells Signaling To Be Phagocytosed
News summary: Mammalian cells that are infected with virus or bacteria put out signals inducing other cells to phagocytose (i.e. eat) them alive, thereby limiting infection. This suggests that infections could be reduced by stimulating such phagocytosis.
Birkle, T. and Brown, G.C., 2021. I’m Infected, Eat Me! Innate Immunity Mediated by Live, Infected Cells Signaling To Be Phagocytosed. Infection and Immunity, 89(5).
DOI: 10.1128/IAI.00476-20
Abstract
Innate immunity against pathogens is known to be mediated by barriers to pathogen invasion, activation of complement, recruitment of immune cells, immune cell phagocytosis of pathogens, death of infected cells, and activation of the adaptive immunity via antigen presentation. Here, we propose and review evidence for a novel mode of innate immunity whereby live, infected host cells induce phagocytes to phagocytose the infected cell, thereby potentially reducing infection. We discuss evidence that host cells, infected by virus, bacteria, or other intracellular pathogens (i) release nucleotides and chemokines as find-me signals, (ii) expose on their surface phosphatidylserine and calreticulin as eat-me signals, (iii) release and bind opsonins to induce phagocytosis, and (iv) downregulate don’t-eat-me signals CD47, major histocompatibility complex class I (MHC1), and sialic acid. As long as the pathogens of the host cell are destroyed within the phagocyte, then infection can be curtailed; if antigens from the pathogens are cross-presented by the phagocyte, then an adaptive response would also be induced. Phagocytosis of live infected cells may thereby mediate innate immunity.
Wild-type sTREM2 blocks Aβ aggregation and neurotoxicity, but the Alzheimer's R47H mutant increases Aβ aggregation
News summary: A protein, sTREM2, appears to protect against Alzheimer’s disease by blocking the aggregation and neurotoxicity of amyloid beta. This suggests that the disease might be delayed by increasing sTREM2 levels in the brain.
Vilalta, A., Zhou, Y., Sevalle, J., Griffin, J.K., Satoh, K., Allendorf, D.H., De, S., Puigdellívol, M., Bruzas, A., Burguillos, M.A. and Dodd, R.B., 2021. Wild-type sTREM2 blocks Aβ aggregation and neurotoxicity, but the Alzheimer's R47H mutant increases Aβ aggregation. Journal of Biological Chemistry, 296.
DOI: 10.1016/j.jbc.2021.100631
Abstract
TREM2 is a pattern recognition receptor, expressed on microglia and myeloid cells, detecting lipids and Aβ and inducing an innate immune response. Missense mutations (e.g., R47H) of TREM2 increase risk of Alzheimer's disease (AD). The soluble ectodomain of wild-type TREM2 (sTREM2) has been shown to protect against AD in vivo, but the underlying mechanisms are unclear. We show that Aβ oligomers bind to cellular TREM2, inducing shedding of the sTREM2 domain. Wild-type sTREM2 bound to Aβ oligomers (measured by single-molecule imaging, dot blots, and Bio-Layer Interferometry) inhibited Aβ oligomerization and disaggregated preformed Aβ oligomers and protofibrils (measured by transmission electron microscopy, dot blots, and size-exclusion chromatography). Wild-type sTREM2 also inhibited Aβ fibrillization (measured by imaging and thioflavin T fluorescence) and blocked Aβ-induced neurotoxicity (measured by permeabilization of artificial membranes and by loss of neurons in primary neuronal–glial cocultures). In contrast, the R47H AD-risk variant of sTREM2 is less able to bind and disaggregate oligomeric Aβ but rather promotes Aβ protofibril formation and neurotoxicity. Thus, in addition to inducing an immune response, wild-type TREM2 may protect against amyloid pathology by the Aβ-induced release of sTREM2, which blocks Aβ aggregation and neurotoxicity. In contrast, R47H sTREM2 promotes Aβ aggregation into protofibril that may be toxic to neurons. These findings may explain how wild-type sTREM2 apparently protects against AD in vivo and why a single copy of the R47H variant gene is associated with increased AD risk.
The Phagocytic Code Regulating Phagocytosis of Mammalian Cells
News summary: There are now known to be about 50 different signals regulating whether one cell eats (phagocytoses) another, and we review here how these signals function as a code determining which cells are eaten in which conditions.
Cockram, T.O., Dundee, J.M., Popescu, A.S. and Brown, G.C., 2021. The phagocytic code regulating phagocytosis of mammalian cells. Frontiers in Immunology, 12.
DOI: 10.3389/fimmu.2021.629979
Abstract
Mammalian phagocytes can phagocytose (i.e. eat) other mammalian cells in the body if they display certain signals, and this phagocytosis plays fundamental roles in development, cell turnover, tissue homeostasis and disease prevention. To phagocytose the correct cells, phagocytes must discriminate which cells to eat using a 'phagocytic code' - a set of over 50 known phagocytic signals determining whether a cell is eaten or not - comprising find-me signals, eat-me signals, don't-eat-me signals and opsonins. Most opsonins require binding to eat-me signals - for example, the opsonins galectin-3, calreticulin and C1q bind asialoglycan eat-me signals on target cells - to induce phagocytosis. Some proteins act as 'self-opsonins', while others are 'negative opsonins' or 'phagocyte suppressants', inhibiting phagocytosis. We review known phagocytic signals here, both established and novel, and how they integrate to regulate phagocytosis of several mammalian targets - including excess cells in development, senescent and aged cells, infected cells, cancer cells, dead or dying cells, cell debris and neuronal synapses. Understanding the phagocytic code, and how it goes wrong, may enable novel therapies for multiple pathologies with too much or too little phagocytosis, such as: infectious disease, cancer, neurodegeneration, psychiatric disease, cardiovascular disease, ageing and auto-immune disease.
Microglial phagocytosis of neurons in neurodegeneration, and its regulation
News summary: We review how microglial phagocytosis of neurons is regulated, and the evidence that microglial phagocytosis of neurons contributes to the neuronal loss in neurodegenerative diseases.
Butler, C.A., Popescu, A.S., Kitchener, E.J., Allendorf, D.H., Puigdellívol, M. and Brown, G.C., 2021. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. Journal of neurochemistry, 158(3).
DOI: 10.1111/jnc.15327
Abstract
There is growing evidence that excessive microglial phagocytosis of neurons and synapses contributes to multiple brain pathologies. RNA-seq and genome-wide association (GWAS) studies have linked multiple phagocytic genes to neurodegenerative diseases, and knock-out of phagocytic genes has been found to protect against neurodegeneration in animal models, suggesting that excessive microglial phagocytosis contributes to neurodegeneration. Here, we review recent evidence that microglial phagocytosis of live neurons and synapses causes neurodegeneration in animal models of Alzheimer's disease and other tauopathies, Parkinson's disease, frontotemporal dementias, multiple sclerosis, retinal degeneration and neurodegeneration induced by ischaemia, infection or ageing. We also review factors regulating microglial phagocytosis of neurons, including: nucleotides, frackalkine, phosphatidylserine, calreticulin, UDP, CD47, sialylation, complement, galectin-3, Apolipoprotein E, phagocytic receptors, Siglec receptors, cytokines, microglial epigenetics and expression profile. Some of these factors may be potential treatment targets to prevent neurodegeneration mediated by excessive microglial phagocytosis of live neurons and synapses.
Sialylation acts as a checkpoint for innate immune responses in the central nervous system
News summary: We review how sialic acid on the surface of neurons and microglia regulates inflammation and phagocytosis in the brain.
Klaus, C., Liao, H., Allendorf, D.H., Brown, G.C. and Neumann, H., 2021. Sialylation acts as a checkpoint for innate immune responses in the central nervous system. Glia, 69(7).
DOI: 10.1002/glia.23945
Abstract
Sialic acids are monosaccharides that normally terminate the glycan chains of cell surface glyco-proteins and -lipids in mammals, and are highly enriched in the central nervous tissue. Sialic acids are conjugated to proteins and lipids (termed "sialylation") by specific sialyltransferases, and are removed ("desialylation") by neuraminidases. Cell surface sialic acids are sensed by complement factor H (FH) to inhibit complement activation or by sialic acid-binding immunoglobulin-like lectin (SIGLEC) receptors to inhibit microglial activation, phagocytosis, and oxidative burst. In contrast, desialylation of cells enables binding of the opsonins C1, calreticulin, galectin-3, and collectins, stimulating phagocytosis of such cells. Hypersialylation is used by bacteria and cancers as camouflage to escape immune recognition, while polysialylation of neurons protects synapses and neurogenesis. Insufficient lysosomal cleavage of sialylated molecules can lead to lysosomal accumulation of lipids and aggregated proteins, which if excessive may be expelled into the extracellular space. On the other hand, desialylation of immune receptors can activate them or trigger removal of proteins. Loss of inhibitory SIGLECs or FH triggers reduced clearance of aggregates, oxidative brain damage and complement-mediated retinal damage. Thus, cell surface sialylation recognized by FH, SIGLEC, and other immune-related receptors acts as a major checkpoint inhibitor of innate immune responses in the central nervous system, while excessive cleavage of sialic acid residues and consequently removing this checkpoint inhibitor may trigger lipid accumulation, protein aggregation, inflammation, and neurodegeneration.